



Drug Safety & Pharmacovigilance professionals have long awaited consistency in the manner in which the US Food and Drug Administration (FDA) accepts Individual Case Safety Report (ICSR) submissions. For years, the industry has navigated different submission procedures for FDA compared with other regions, as well as differences between Investigational New Drug (IND) reporting and post-marketing pharmacovigilance reporting. The FDA has been working towards harmonization of ICSR submissions and the final guidance is here! FDA will require that both premarket and post-market safety reports be submitted electronically in the ICH E2B(R3) format, the format that is currently the standard in the EU. Let us explore the current FDA practices, the need for updated processes, timelines for implementation, and what pharmacovigilance professionals need to do to prepare for the change.
Current Processes for Submitting ICSRs to FDA
Expedited ICSRs submitted under an IND:
Information received on a safety event is entered into the safety database and a MedWatch (Form 3500 A) PDF is generated. This document, together with a cover letter and Form 1571, is submitted to the IND.
This process is time consuming and imposes certain limitations. Regulatory Affairs review and approval is required between the safety database output and the submission. For multinational studies, this means there is an extra step for US reporting that is not included in reporting in the EU. This is inefficient for the Sponsor. In addition, FDA’s review time is lengthened because the PDF format does not allow for easy data analysis and visualization.
Post-marketing ICSRs
ICH E2B XML files are submitted through the FDA Adverse Event Reporting System (FAERS), currently in E2B(R2) format.
This format is no longer aligned globally; many countries in Europe, Asia and the rest of the world have already transitioned to E2B(R3). This imposes issues in backward and forward compatibility between E2B(R2) and E2B(R3) formats, which can cause validation errors and is an example of inefficiency caused by the need for an additional report for global products.
New Process for Submitting ICSRs to FDA After Implementation of E2B(R3)
According to the final guidance, XML files will be submitted through FAERS in ICH E2B(R3) format for both IND and post-marketing ICSRs. Most of the above-mentioned limitations of the current processes will be resolved while remaining compliant with US regulations stipulated in 21 CFR 312.32.(c)(1)(v). Of note, there will be some exceptions to the new process (findings from other studies, findings from animal or in vitro testing, and Increased rate of occurrence of serious suspected adverse reactions), as noted in 21 CFR 312.32.(c)(1)(iii-iv).
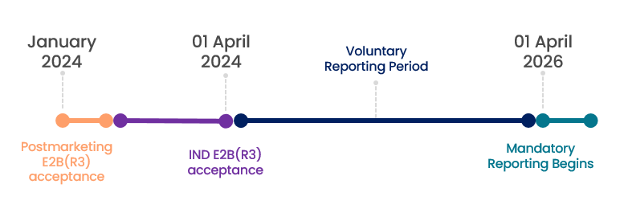
Timeline for Implementation
FDA posted their final guidance on 01 April 2024. From that date, reporting using the new process will be voluntary for 24 months and mandatory starting 01 April 2026.
Preparing Your Technology and Procedures
Sponsors have until 01 April 2026 to prepare their systems and procedures before the new format becomes mandatory. It is advisable that pharmacovigilance teams start planning and modifying technology and procedures now, as this will take substantial time and effort. It is a good practice to start submissions in the new format during the Voluntary Reporting Period to allow time for modifications to processes before Mandatory Reporting begins. Note that once a Sponsor makes the first E2B(R3) submission in the Voluntary Period, they will not be allowed to revert to legacy reporting methods.
The FDA has published many resources to prepare Sponsors for the change. I have provided a list of resources at the end of this blog. Sponsors must learn the technology specifications, determine whether an upgrade or update to the safety database is needed, create an electronic submissions gateway (ESG) and WebTrader account, if they do not already exist, update SOPs, processes, manuals, and training materials, and perform validation testing. Challenges and rework should be expected and time allowed in the plan to cover these issues.
How Will the New Process Improve Safety Surveillance?
With the implementation of the new format and submission process, safety data from IND studies and post-marketing reports will be housed in one source. This harmonization of data sources will improve FDA reviews of clinical trial and post-marketing data across a product and product class. This will greatly enhance efficiency of signal detection and FDA safety and quality reviews.
How Will the New Process Benefit PROMETRIKA’s Sponsors?
The new system makes it straightforward, even advisable, for Sponsors to delegate submission of IND and pharmacovigilance safety reports to a single pharmacovigilance services provider. Currently, PROMETRIKA Pharmacovigilance processes both pre- and post-marketing safety reports for our clients. However, while we can send post-marketing reports directly to FAERS on behalf of our Sponsors, the Sponsor must retain FDA submission responsibilities for IND submissions unless PROMETRIKA is also responsible for IND maintenance. For IND submissions, we prepare the MedWatch PDF and send it to the Sponsor for submission. Implementing E2B(R3) in a premarket setting will allow PROMETRIKA to submit all safety reports on behalf of our Sponsors, directly from the safety database using FDA’s ESG. This will simplify global reporting and give our Sponsors confidence in the accuracy and consistency of safety data.
In summary, converting your processes to meet the updated FDA requirements will be a lot of work, but in the end, it will be worth it for everyone!
References:
21 CFR 312.32
Good Clinical Practice & Pharmacovigilance Symposium, February 15, 2024: FDA Adverse Event Reporting System (FAERS) Updates Suranjan De, MS, MBA Deputy Director of the Regulatory Science Staff | OSE | FDA
FDA Adverse Event Reporting System (FAERS) Electronic Submissions –Web page: FDA Adverse Event Reporting System (FAERS) Electronic Submissions | FDA
FDA Regional Implementation Guide for E2B(R3) Electronic Transmission of Individual Case Safety Reports for Drug and Biological Products (Aug 2022): FDA Regional Implementation Guide for E2B(R3) Electronic Transmission of Individual Case Safety Reports for Drug and Biological Products | FDA
FDA E2B(R3) Core and Regional Data Elements and Business Rules v1.6 (Jan 2024): FDA Adverse Event Reporting System (FAERS) Electronic Submissions – E2B(R3) Standards | FDA
FDA E2B(R3) Forward Compatible Rules (Apr 2022): https://www.fda.gov/media/157993/download
FDA ICSR XML Instances (Sep 2023): https://www.fda.gov/media/157983/download
Electronic Submission of IND Safety Reports - Technical Conformance Guide (Apr 2022): Electronic Submission of IND Safety Reports Technical Conformance Guide | FDA
Electronic Submission of Expedited Safety Reports from IND-Exempt BA/BE Studies - Draft Guidance for Industry (Aug 2022): Electronic Submission of Expedited Safety Reports From IND-Exempt BA/BE Studies Guidance for Industry | FDA
Create an ESG account if you do not already have one: Setting up a WebTrader Account Checklist | FDA
Validate E2B(R3) XML files generated from your Safety Database: https://faers2-validator.preprod.fda.gov/LSMV/Validator
Providing Regulatory Submissions in Electronic Format: IND Safety Reports: Guidance for Industry April 2024

