



In December 2025, FDA published two final guidance documents regarding safety reporting requirements for investigational agents – one geared to Sponsor requirements for INDs and bioavailability/bioequivalence studies and the other focused on Investigator requirements for drug and device studies.
The first guidance, Sponsor Responsibilities – Safety Reporting Requirements and Safety Assessment for IND and Bioavailability/Bioequivalence Studies finalizes the draft guidance issued in June 2021 and is applicable to both drugs and biologics. For the purposes of this blog, I will refer to drugs and biologics as investigational agents. The second guidance, Investigator Responsibilities – Safety Reporting for Investigational Drugs and Devices: Guidance for Industry, covers Investigator responsibilities for safety reporting for investigational agents and devices.
DEFINITIONS
Both guidance documents start with definitions to make sure that we are all on the same page and using the correct terms.
A very useful set of definitions highlighted in these guidance documents deal with the terms “expected versus anticipated.” Expected refers to events that are listed in the Investigator Brochure (IB) or Reference Safety Information (RSI) and as a result have been reported as associated with the use of the investigational agent. Anticipated events are those which have not been reported as associated with the use of the investigational agent but would be anticipated based on the pharmacological class, patient population and/or disease factors.
INVESTIGATOR GUIDANCE
In my view, it is nice to have a guidance specifically directed to Investigators and their roles and responsibilities regarding safety in clinical trials. This guideline outlines Investigator reporting requirements to Sponsors and IRBs. We all know that Investigators are required to report serious adverse events (SAEs) expeditiously and it is nice to see that FDA agrees and states that this should be “immediately,” which generally translates to within 24 hours. The guidance also states that Investigators should read the safety reports that they receive from Sponsors and report the information to IRBs in accordance with IRB policy.
SPONSOR GUIDANCE
Most of this blog will focus on this guidance for Sponsors.
By way of introduction, it is important to note that there is a difference between the IND safety reporting regulation under 21CFR 312.32 and the ICH E2A guidance with respect to who is responsible for making the final judgment regarding causality for safety reporting purposes. While both guidance documents recommend that the Investigator provide a causality assessment, for FDA reporting, it is the Sponsor assessment that should be used. The ICH E2A guidance recommends reporting if either the Investigator or the Sponsor report a positive causality assessment. The reason that FDA has made this distinction is that the Sponsor has more information upon which to base causality decisions as the Sponsor generally is responsible for the global safety database.
Much of the Sponsor guidance focuses on aggregate reporting and provides very useful information to consider in doing these analyses. Thinking back to when the initial guidance on this topic (Safety Reporting Requirements for INDs and BA/BE Studies [December 2012]) was issued, we in Industry spent a great deal of time trying to determine how best to meet the aggregate reporting requirements. I was very happy to see that FDA recognizes and understands the difficulties in conducting these required safety assessments and determining when an aggregate report is appropriate. One of the big take-home messages, from my perspective, is the recommendation that the Sponsor develop a Safety Surveillance Plan for each investigational agent. With this plan in place, regularly scheduled safety reviews are conducted and documented.
Maintaining trial integrity is also highlighted, including appropriate procedures and controls to prevent unblinding. Sponsor personnel conducting unblinded safety reviews should not participate in the conduct or analysis of aggregate reviews. If a Data Monitoring Committee (DMC) is involved in this type of review, the guidance recommends that the DMC Charter specify this.
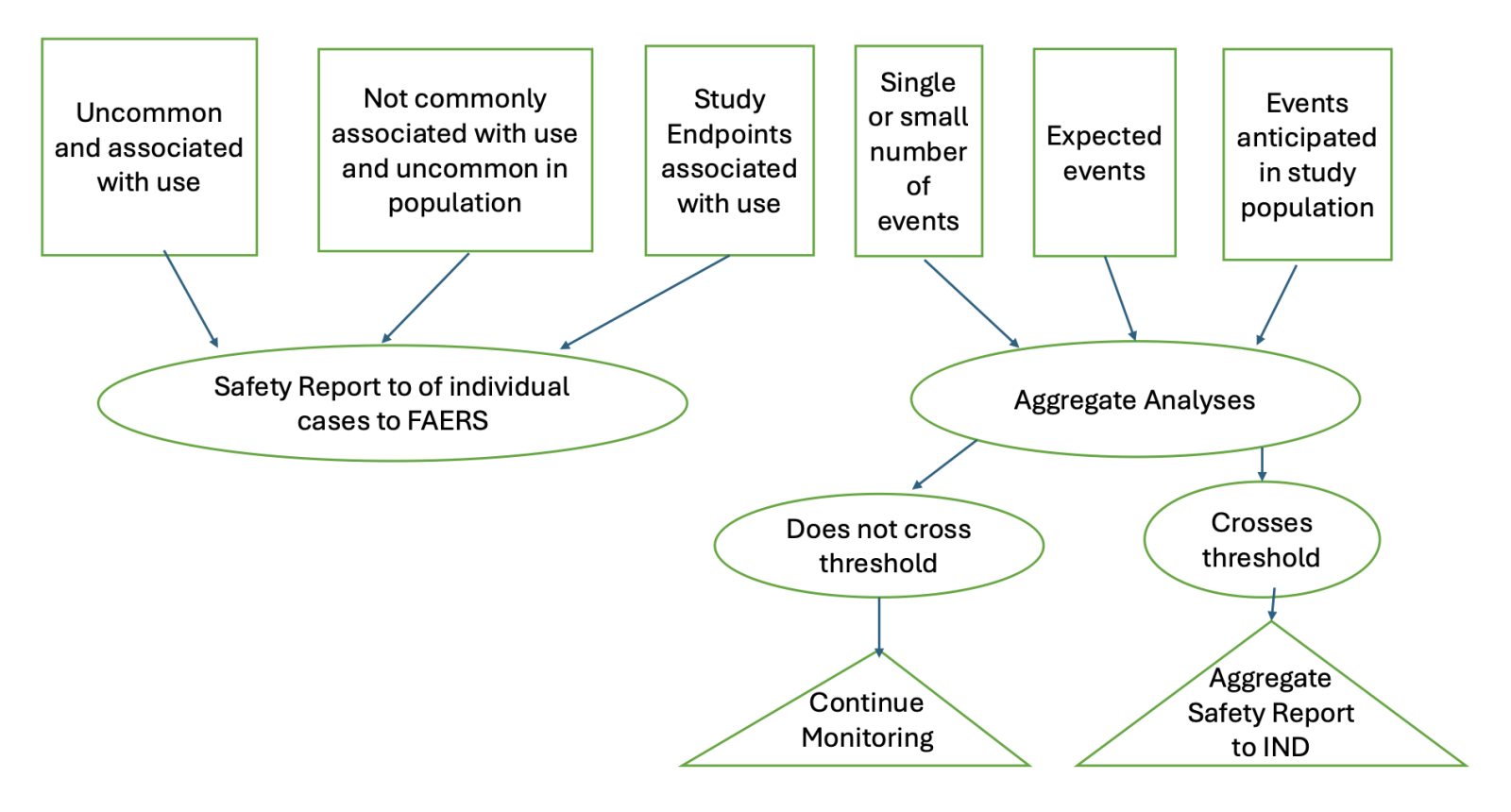
Safety reports based on aggregate analyses are submitted to the IND, not directly to the FDA Adverse Event Reporting System (FAERS), as is required for individual safety reports. When one thinks about this, it makes perfect sense as an aggregate safety report will be accompanied by additional documentation such as changes in the protocol, ICF, RSI, and/or an Investigator Brochure update.
When IND safety reports based on aggregate data are generated, FDA recommends sending only the summary portion of the IND safety report to all participating investigators. The individual unblinded safety cases can be omitted.
Take home messages regarding analyses of aggregate data and reporting decisions outlined in the guidance are as follows:
- Single or Small Number of Cases
It is often difficult to make a causal determination based on a single or small number of cases. This will be especially true during the early stages of clinical development and this underscores the need for routine aggregate analyses and documentation of the results and conclusions.
- Increase in the Number of Expected Cases
Clinically significant increase in the number of expected events can result in an aggregate report and a change in the RSI.
- Events that Do Not Require Aggregate Analysis and Should be Reported as Individual Safety Reports to FAERS
-
- Single Occurrences that are Uncommon and Known to be Strongly Associated with Drug Exposure
-
Stevens-Johnson Syndrome is a good example of this. In these cases, one occurrence is enough for reporting. If this occurs in a blinded study, the blind should be broken prior to reporting. If the event happened in a subject receiving placebo, the event does not need to be reported.
-
-
- Events Not Commonly Associated with Drug Exposure
-
One or more occurrences of an event not commonly associated with drug exposure and uncommon in the population being treated may be a suspected adverse reaction if it occurs in conjunction with the administration of the investigational product, or recurs when the product is administered again. In this instance, a single case may be sufficient for safety reporting.
- Events that Require Aggregate Analyses
Events that are expected in the study population, independent of drug exposure, and not listed in the IB or RSI, meet these criteria. The difficult part is determining when these events would reach the threshold for aggregate reporting. For this, there is guidance on how to approach these aggregate analyses and reporting decisions, including background rates in the population, rates of reporting in study arms of a randomized study, and an assessment of whether there is a reasonable possibility that the investigational agent increased the incidence of the event. An increased occurrence of a clinically important expected serious suspected adverse event would also meet these criteria.
- Safety Reports from Study Endpoints
Study endpoints are not expected to be reported as IND safety reports unless there is evidence of a causal relationship. In this guidance, FDA provides a nice example of when IND safety reporting would be appropriate.
“If, however, the death occurred as a result of an anaphylactic reaction that coincided with initial exposure to the drug or was the result of fatal hepatic necrosis, the death must be reported as an individual case in an IND safety report because of the evidence suggesting a causal relationship between the drug and the event.”
STANDARD CODING
Last but not least, is the importance of standard coding, as this is integral in the review and assessment of safety signals.
Below is a flow chart of the scenarios discussed above and the reporting decisions and FDA submissions for safety reports based on individual events as well as aggregate analyses.
Flow Chart: Serious Adverse Event Processing, Aggregate Analysis and Reporting Decisions
We at PROMETRIKA assist our clients with regulatory, clinical development, statistical, pharmacovigilance, and DMC services. Through these activities, we are uniquely positioned to assist our clients with safety services, including direct submission to FAERS of individual cases as well as the design and execution of aggregate analyses.

